THESIS in Microbiology & Immunology
Exploring Poxvirus Genome Packaging using CRISPR-Cas Systems
Fall 2024, Volume 23, Issue 1
Daniel Kuo ’24, Brian Ward*
https://doi.org/10.47761/BNHJ9426
Introduction:
The compaction and packaging of genetic material are intricate and important processes shared among all three domains of life and viruses (1). The specific mechanisms by which viruses package their genome vary from species to species. For most DNA and RNA viruses with small genomes (<20 kb), an energy-independent system is used, where the capsid is assembled around the genome (1). In contrast, viruses with larger genomes tend to use energy-dependent systems, where ATP-driven motors pump the genome into a preformed capsid (1). Furthermore, many viruses use packaging signals to selectively package viral RNA or DNA as opposed to host RNA or DNA. These signals are present in several notable large double-stranded DNA viruses. For example, adenovirus genomes contain an AT-rich packaging domain that interacts with viral and cellular proteins to mediate genome packaging (2). Additionally, herpesvirus genomes contain two sequence motifs named pac1 and pac2 that are involved in concatemer cleavage and genome packaging (3). While poxviruses are also large double-stranded DNA viruses, similar packaging motifs have yet to be identified.
The Poxviridae family consists of large DNA viruses that replicate entirely in the cytoplasm of vertebrate or invertebrate cells (4). Vertebrate poxviruses are grouped into six genera, with the Orthopoxvirus genus being the most extensively studied (4, 5). Several notable viruses belong to this genus, including mpox, vaccinia virus (VACV), and variola virus, which is the causative agent of smallpox (6). VACV serves as the prototypic member of the Poxviridae family, and the extensive study of this virus has contributed to several key developments, such as the mpox and smallpox vaccines (6).
Transcription of the VACV genome occurs in a temporally-regulated fashion, divided into early, intermediate, and late stages (7). Early genes, which are expressed shortly after infection and before uncoating, encode proteins involved in host immune evasion, viral DNA synthesis, and intermediate gene expression (4). DNA replication is required for intermediate and late gene expression; intermediate genes encode enzymes and factors required for late gene expression (4,7).
A common characteristic of poxvirus genomes is the presence of AT-rich inverted terminal repeats (ITRs) which are identical, but oppositely oriented sequences found at the two ends of the genome (4). For VACV specifically, the entire genome is a 195-kb linear duplex that is AT-rich (~67%) (7). The genes contain 5’ and 3’ untranslated regions, lack introns, and can be encoded on either strand of the genome (7). ITRs of about 10 kb are present at the distal ends of the genome to link the two strands into a covalently closed molecule (7). After early gene transcription, uncoating inside the cell takes place which releases the genome. Thus replication undergoes in discrete cytoplasmic structures called replication factories (7). Nascent genomes are then delivered into immature virions (IVs) before the membrane closes, forming IVs with nucleoids that further mature to form infectious mature virions (MVs) (7).
The replication and packaging of the VACV genome remains poorly understood. Since no in vitro replication system has been established, models of poxvirus replication remain speculative. Most discussions focus on a self-priming model where a nick is introduced in the ITRs (7), providing a 3’-OH that can serve as the site of initiation for DNA synthesis. The self-priming model labels infected cells with [3H]thymidine and observes that the radiolabel was first incorporated within the ITR (8). Furthermore, prior research has demonstrated that the ITR is involved in replication and certain VACV proteins (I1, I6, K4) interact with the ITR (9). However, it remains unclear whether these proteins are involved in DNA replication or marking the genome for encapsidation (9).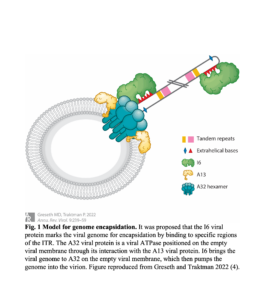
A proposed model for genome packaging, shown in Figure 1, posits that the I6 viral protein marks the viral genome for encapsidation by binding the genomic termini, which is AT-rich, contains extra helical bases, and forms hairpin structures (7). Afterward, I6 brings the viral genome to the A32 viral protein, a viral ATPase positioned at the empty viral membrane to pump the genome into the virion (7). While packaging signals have been identified in other double-stranded DNA viruses like herpes simplex virus type 1 and adenovirus, similar sequences have not been identified in poxviruses (8).To explore the location of packaging signals, clustered regularly interspaced short palindromic repeats and CRISPR-associated protein (CRISPR-Cas) systems, in conjugation with fluorescent protein reporters, can be utilized to fragment the VACV genome and determine packaged fragments.
Many archaea and bacteria utilize CRISPR-Cas systems composed of Cas effector proteins and CRISPR RNAs (crRNAs) to defend against attacks by foreign mobile genetic elements (MGEs) like plasmids or phages (10, 11). CRISPR-Cas systems work by their proteins forming an adaptation complex that recognizes the protospacer sequence on invading DNA/RNA via the protospacer adjacent motif (PAM) (10, 11). This sequence is incorporated into the CRISPR array, an area of the prokaryotic genome composed of short and conserved repetitive sequences, called repeats, between unique spacer sequences. The CRISPR array is transcribed into a long precursor crRNA (pre-crRNA) and processed into a shorter crRNA molecule that guides the Cas endonuclease to the target sequence for cleavage (10, 11). CRISPR-Cas systems are divided into two groups that are subdivided into six types (I to VI) based on their effector nucleases: Class 1 systems use several Cas proteins while class 2 systems use a single-component Cas protein (10). Class 2 systems (types II, V, and VI) comprise approximately 10% of all identified CRISPR-Cas loci and are found almost exclusively in bacteria (11).
Two notable examples of Class 2 systems are Cas12a and Cas9. Cas12a is a type V Cas protein that forms a ribonucleoprotein complex with a single crRNA molecule (11). Unlike type II systems which utilize bacterial RNase III with trans-activating crRNA (tracrRNA) to process the pre-crRNA, Cas12a has endoribonuclease activity and can process its pre-crRNA into mature crRNA without r tracrRNA (11). This gives it an advantage over CRISPR-Cas systems that require tracrRNA, like Cas9, as the system’s design can be simplified due to needing one less component. Additionally, for the expression of CRISPR-Cas systems, for genome editing in plants and animals, the RNA polymerase type III U6 promoter is utilized to express precise, uncapped, non-polyadenylated crRNA molecules. The ability of the Cas12a endonuclease to process its pre-crRNA into crRNA means that crRNA expression can come from a wide range of promoters, including viral ones. Furthermore, it has been demonstrated that Cas12a can generate a staggered DNA double-stranded break with a 4 or 5 nucleotide 5’ overhang on target DNA preceded by a short T-rich PAM (10). This gives CRISPR-Cas12a an advantage over other CRISPR-Cas systems for targeting sequences in the orthopoxvirus genome that have an AT-rich nature by providing an increased number of targets. The specific PAM sequence can vary between species. For example, the PAM for LbCas12a (Lachnospiraceae bacterium) and AsCas12a (Acidaminococcus) has the sequence 5’-TTTN-3′; for FnCas12a (Francisella novicida), the PAM sequence 5’-TTN-3’ is located upstream of the 5′ end of the non-target strand (11). FnCas12a has been demonstrated to cut the 18th base after the PAM on the non-targeted (+) strand and the 23rd base on the targeted (-) strand (10). This break can be repaired through nonhomologous end joining (NHEJ), typically leading to short insertion/deletion (indels) near the cut site that inactivates protein-coding genes by inducing frameshifts (10,12). If template DNA homologous to the break site is present, this double-stranded break can also be repaired through homology-directed repair (HDR) (11). HDR allows exogenous sequences with homology to the cut site to be introduced into the genome, allowing for the incorporation of specific sequences at a target site (12).
Cas9 is a type II Cas protein that forms a ribonucleoprotein complex with two noncoding RNAs, the crRNA and tracrRNA (12). These two RNA molecules can be fused into a single guide RNA (sgRNA) for genome editing (12). The Cas9/sgRNA complex then binds double-stranded DNA sequences that match the first 17-20 bases of the sgRNA only if the sequence is followed by a PAM, which has the sequence 5’-NGG-3’ for SpCas9 (Streptococcus pyogenes) (12). Once bound, the Cas9 endonuclease cleaves both DNA strands 3 bases upstream of the PAM, resulting in a blunt end DNA double-stranded break (12). Like CRISPR-Cas12, this break can be repaired through NHEJ, typically leading to indels near the cut site that inactivates protein-coding genes by inducing frameshifts (12). Since VACV early gene expression occurs before uncoating, early gene ORFs may be sequestered from the CRISPR-Cas system by the capsid and as a result, cannot be accessed before early gene expression. While the VACV genome–located in the cytoplasm–can be efficiently cut with CRISPR-Cas9, prior research did not observe NHEJ repair suggesting that the viral genome can be cleaved into smaller fragments (13). We hypothesize that bona fide packaging signals exist within the VACV genome. To map these signals, we initially designed a CRISPR-Cas12a system, but ultimately developed a CRISPR-Cas9 system to cut the VACV genome into smaller fragments. By adding different fluorescent protein reporters to each fragment, it will then be determined which, if any, of these smaller fragments are packaged. Reported here is the progress made toward developing a FnCas12a system and SpCas9 system that cleaves the Emerald green fluorescent protein (EGFP) gene inserted in the VACV genome.
Results:
No observed differences in EGFP expression between treatment with the CRISPR-Cas12a system and the negative control.
Initially, we sought to observe the cutting of the EGFP gene in the VACV genome by a virally-expressed CRISPR-Cas12a system. Two crRNA, or guide RNA (gRNA), oligonucleotides targeting the 3’ and 5’ regions of the EGFP gene were designed based on Cpf1 parameters described in Zetsche et al. 2015 (10). These oligonucleotides were cloned into the pMiniT 2.0 backbone under the control of the I1 viral promoter and termed p3gRNA (3’ gRNA) and p5gRNA (5’ gRNA) (Table 1). The pCas12a is the plasmid that expresses the FnCas12a endonuclease, with expression of both gRNAs and the FnCas12a driven by the I1 viral promoter, an intermediate VACV promoter. The 5’ crRNA binds starting at nucleotide 45 and the 3’ crRNA binds starting at nucleotide 607 on the EGFP open reading frame (ORF), with cleavage generating a 5 nucleotide 5’ overhang with some complementarity (Figure 2).
| Table 1: gRNA oligonucleotide sequences and corresponding plasmids | ||
| Oligonucleotide Name | Sequence (5’-3’) | Plasmid Name |
| EGFP 3’ gRNA Rev | CTGAGCACCCAGTCCGCCCTGAGATCTACAACAGTAGAAATTTAAGTTCACC | p3gRNA |
| EGFP 5’ gRNA Rev | CCTGGTCGAGCTGGACGGCGACGATCTACAACAGTAGAAATTTAAGTTCACC | p5gRNA |
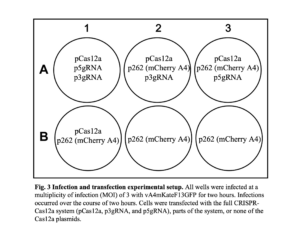
 HeLa cells were infected with a recombinant VACV that expresses red fluorescent protein (RFP) tagged A4 proteins and EGFP-tagged F13 proteins (vA4mKateF13GFP). It was expected that when the EGFP ORF was fragmented, little to no EGFP would be expressed compared to the control cells. Regardless, infected cells should express RFP. The cells were then transfected with plasmids as shown below in Figure 3. Well A1 contained the following plasmids: pCas12a for FnCas12a expression, p3gRNA for the 3’ gRNA expression, and p5gRNA for the 5’ gRNA expression. Expression of these components was driven by the I1 intermediate viral promoter. All components necessary for the FnCas12a protein to cleave the EGFP gene were present in wells A1, A2, and A3 but showed no difference in EGFP expression compared to the control wells (wells B1, B2, B3), which lacked plasmids encoding elements of the CRISPR-Cas12a system.
HeLa cells were infected with a recombinant VACV that expresses red fluorescent protein (RFP) tagged A4 proteins and EGFP-tagged F13 proteins (vA4mKateF13GFP). It was expected that when the EGFP ORF was fragmented, little to no EGFP would be expressed compared to the control cells. Regardless, infected cells should express RFP. The cells were then transfected with plasmids as shown below in Figure 3. Well A1 contained the following plasmids: pCas12a for FnCas12a expression, p3gRNA for the 3’ gRNA expression, and p5gRNA for the 5’ gRNA expression. Expression of these components was driven by the I1 intermediate viral promoter. All components necessary for the FnCas12a protein to cleave the EGFP gene were present in wells A1, A2, and A3 but showed no difference in EGFP expression compared to the control wells (wells B1, B2, B3), which lacked plasmids encoding elements of the CRISPR-Cas12a system.
 No difference in the number of cells fluorescing RFP and EGFP was observed between well A1 and the negative control wells, which suggested that the FnCas12a system was not cutting the EGFP in a measurable way. We theorized that this could be related to factors such as the high multiplicity of infection (MOI), the CRISPR-Cas protein not expressing, the crRNAs not targeting the EGFP ORF, or the crRNAs not being expressed. A high MOI can induce antiviral responses and cytotoxicity, potentially causing undesirable effects that may interfere with the CRISPR-Cas system. Additionally, the genome’s replication rate may outpace the CRISPR-Cas12a system’s ability to locate and cleave it. This means cutting the template genome before replication will result in fewer replicated genomes being made, compared to fragmenting it after several rounds of replication have already taken place. Moreover, FnCas12a-mediated cleavage requires the expression of both the FnCas12a nuclease and the crRNAs. Without proper expression of the FnCas12a endonuclease, it is not expected that DNA cleavage will be observed. Additionally, without proper expression of the gRNAs, DNA cleavage will be absent because the FnCas12a endonuclease requires the gRNAs to target a specific DNA sequence. If either of these components is not properly expressed, the FnCas12a system will not be able to cut the EGFP gene.
No difference in the number of cells fluorescing RFP and EGFP was observed between well A1 and the negative control wells, which suggested that the FnCas12a system was not cutting the EGFP in a measurable way. We theorized that this could be related to factors such as the high multiplicity of infection (MOI), the CRISPR-Cas protein not expressing, the crRNAs not targeting the EGFP ORF, or the crRNAs not being expressed. A high MOI can induce antiviral responses and cytotoxicity, potentially causing undesirable effects that may interfere with the CRISPR-Cas system. Additionally, the genome’s replication rate may outpace the CRISPR-Cas12a system’s ability to locate and cleave it. This means cutting the template genome before replication will result in fewer replicated genomes being made, compared to fragmenting it after several rounds of replication have already taken place. Moreover, FnCas12a-mediated cleavage requires the expression of both the FnCas12a nuclease and the crRNAs. Without proper expression of the FnCas12a endonuclease, it is not expected that DNA cleavage will be observed. Additionally, without proper expression of the gRNAs, DNA cleavage will be absent because the FnCas12a endonuclease requires the gRNAs to target a specific DNA sequence. If either of these components is not properly expressed, the FnCas12a system will not be able to cut the EGFP gene.
No observable difference in EGFP expression and the number of progeny virions produced at different MOIs.
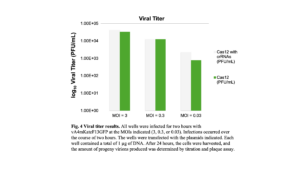
The high MOI of the previous experiment may induce cellular responses that interfere with the CRISPR-Cas12a system. To optimize the MOI and determine how it impacts the CRISPR-Cas system, HeLa cells were infected at different MOIs (3, 0.3, or 0.03) and transfected with either the full FnCas12a system (pCas12a, p3gRNA, p5gRNA) or only the FnCas12a effector protein (pCas12a). Again, there was no clear difference in EGFP expression between the wells transfected with the complete FnCas12a system and the wells that lacked the crRNAs. Thus, comparing the fluorescent proteins alone may not be an adequate way of assessing if the CRISPR-Cas12a system is working because the fluorescent proteins may not be sensitive enough. Double-stranded breaks in the viral genome should not be repaired because NHEJ was not previously observed after the VACV genome was cleaved with CRISPR-Cas9 (13). If the viral genome is not repaired and remains fragmented before encapsidation, fewer infectious progeny virions may be produced since the genes required for an efficient replication cycle will be absent if only a fragment of the genome is packaged. As a result, the progeny virions were quantified by titration to determine the efficiency of the viral replication process (Figure 4). It was expected that infected cells transfected with the full Cas12a system should produce fewer progeny virions compared to the negative control group. However, no significant difference between the number of progeny virions was observed between the experimental and control groups for all MOIs, suggesting that the CRISPR-Cas12a system did not cut the viral genome (Figure 4).
 FnCas12a is expressed in infected cells.
FnCas12a is expressed in infected cells.
The CRISPR-Cas12a system requires two components: the FnCas12a endonuclea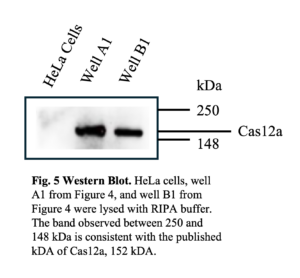 se and the gRNAs. If the CRISPR-Cas protein was not expressed, there would be no EGFP gene cleavage and the observed similarity in the PFU concentration between the experimental and control groups. To confirm the FnCas12a protein expression, a western blot was performed using an antibody that recognized the hemagglutinin (HA) epitope on FnCas12a after lysing the cells in the previous experiment, shown in Figure 4. The western blot revealed a band at approximately 152 kDa, consistent with the predicted size of the FnCas12a protein (Figure 5).
se and the gRNAs. If the CRISPR-Cas protein was not expressed, there would be no EGFP gene cleavage and the observed similarity in the PFU concentration between the experimental and control groups. To confirm the FnCas12a protein expression, a western blot was performed using an antibody that recognized the hemagglutinin (HA) epitope on FnCas12a after lysing the cells in the previous experiment, shown in Figure 4. The western blot revealed a band at approximately 152 kDa, consistent with the predicted size of the FnCas12a protein (Figure 5).
No observed difference in EGFP expression when gRNAs were shortened with a restriction enzyme and infected/transfected in the presence of AraC.
If the FnCas12a endonuclease was expressed but not cleaving, it could indicate that the crRNAs may not be expressed. The CRISPR array is transcribed as a continuous transcript (pre-crRNA), which is then processed into mature crRNAs, 42 to 44 nucleotides in length, by the Cas12a nuclease without the need for tracrRNA (10). The Cas12a nuclease has intrinsic endoribonuclease activity that enables it to cleave the pre-crRNA directly upstream of a repeat-derived stem-loop structure, as shown in Figure 6 (14).
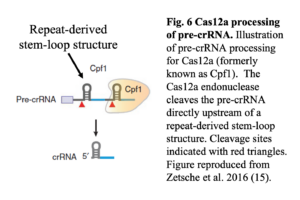 The plasmids that express the 5’ and 3’ crRNAs (p5gRNA and p3gRNA respectively) using the I1 viral promoter do not possess RNA-transcription termination sequences after the crRNA sequences. This can lead to run-on transcription and the formation of a crRNA molecule, which lacks the necessary secondary structure and a 3’ end that may be too long for FnCas12a to effectively process. Without adequately processed crRNA molecules, the FnCas12a protein may not efficiently target and cleave the DNA target. To eliminate run-on transcription and shorten the 3’ end, p3gRNA and p5gRNA were cut with the restriction enzyme NotI, which targets nucleotide 583 on the parental pMiniT 2.0 vector, located immediately downstream from the gRNA insertion site. Transfection with these shortened sequences was compared to transfection with the uncut plasmids and transfection with p331 (irrelevant plasmid). Furthermore, each condition was infected and transfected in the presence or absence of cytosine arabinoside (araC). AraC reversibly inhibits vaccinia virus DNA replication and subsequent morphogenesis, potentially allowing more time for the CRISPR-Cas12a system to cut the VACV genome before replication.
The plasmids that express the 5’ and 3’ crRNAs (p5gRNA and p3gRNA respectively) using the I1 viral promoter do not possess RNA-transcription termination sequences after the crRNA sequences. This can lead to run-on transcription and the formation of a crRNA molecule, which lacks the necessary secondary structure and a 3’ end that may be too long for FnCas12a to effectively process. Without adequately processed crRNA molecules, the FnCas12a protein may not efficiently target and cleave the DNA target. To eliminate run-on transcription and shorten the 3’ end, p3gRNA and p5gRNA were cut with the restriction enzyme NotI, which targets nucleotide 583 on the parental pMiniT 2.0 vector, located immediately downstream from the gRNA insertion site. Transfection with these shortened sequences was compared to transfection with the uncut plasmids and transfection with p331 (irrelevant plasmid). Furthermore, each condition was infected and transfected in the presence or absence of cytosine arabinoside (araC). AraC reversibly inhibits vaccinia virus DNA replication and subsequent morphogenesis, potentially allowing more time for the CRISPR-Cas12a system to cut the VACV genome before replication.
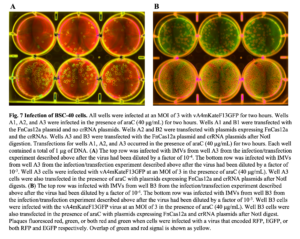
Poxviruses have multiple infectious forms, one being intracellular mature virions (IMVs), which are found inside cells and released via lysis (4). To isolate the IMVs and assess fluorescent protein expression, cells in wells A3 and B3 were lysed (Figure 7). Wells A3 and B3 were transfected with the FnCas12a plasmid and crRNA plasmids after NotI digest. Well A3 was infected in the presence of araC (40 μg/mL) while Well B3 was infected without araC. A plaque assay was performed using BSC-40 cells, a line of African green monkey cells derived from BSC-1 cells that are routinely used for VACV plaque assays, which were then infected with the progeny virions and imaged after 24 hours. These virions encode for EGFP and RFP-tagged proteins. If no EGFP gene cleavage occurs, all plaques will fluoresce green and red. If EGFP gene cleavage and knockout occur, plaques that fluoresce only red but not green will be present. Given the presence of plaques that only fluoresce both red and green, there was no clear indication that the EGFP gene was being cut (Figure 7).
 FnCas12a-mediated cleavage of the EGFP gene was not observed with transient EGFP expression.
FnCas12a-mediated cleavage of the EGFP gene was not observed with transient EGFP expression.
It is possible that the FnCas12a system cannot target the EGFP gene present in recombinant VACV genomes, as the genomes are sequestered in the replication factory. To account for this, HeLa cells were transfected with the CRISPR-Cas12a system and pT7GFP, a plasmid that encodes EGFP under the control of the T7 promoter. pT7GFP expression was driven by the VACV T7 expression system (16).
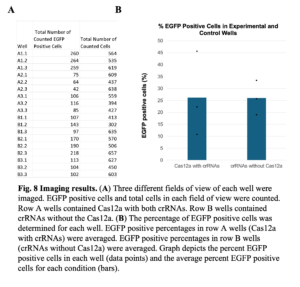
After 24 hours, three different fields of view of each well were imaged. The total number of cells and the total number of EGFP-expressing cells were counted and compared (Figure 8). The percentage of EGFP-expressing cells in the experimental group (FnCas12a with both crRNAs) and the negative control group (crRNAs without FnCas12a) were 26.19% and 26.01%, respectively. No significant difference between these values suggests that there was no significant cleavage of pT7GFP by FnCas12a. Examining the percentage of cells that expressed EGFP for each specific well, A2 had a lower percentage of EGFP-expressing cells compared to the negative control wells. This result would be consistent with the Cas12a endonuclease cutting the EGFP gene, thus reducing EGFP expression.
 PCR amplification of a segment of the EGFP ORF targeted by CRISPR-Cas12a revealed a single band, suggesting that FnCas12a did not cut the gene.
PCR amplification of a segment of the EGFP ORF targeted by CRISPR-Cas12a revealed a single band, suggesting that FnCas12a did not cut the gene.
It is possible that the difference between wells A2 and B2 was obtained through random chance and not a functional CRISPR-Cas12a system. To further analyze our experiment, specific EGFP primers were designed to bind outside the CRISPR-Cas12a cut sites and amplify a region of 714 base pairs between nucleotide 7 and nucleotide 721 of the EGFP coding region via polymerase chain reaction (PCR) (Figure 9). For cells that were transfected with pT7GFP without the Cas12a system, the expected PCR amplicon size was 714 base pairs. For cells transfected with the CRISPR-Cas12a system, Cas12a-mediated double-stranded breaks are repaired with NHEJ which may introduce indels. As a result, the expected PCR amplicon size may be either larger or smaller than 714 base pairs. Since 2 separate sgRNAs were used, it may be possible to simultaneously generate two staggered double-stranded DNA breaks on the plasmid at each cut site. The sgRNAs target sequences are approximately 560 bases apart, meaning that if NHEJ occurs between the two staggered ends of the plasmid, PCR amplification of pT7GFP should produce a distinct band of approximately 560 bases smaller than the uncut EGFP gene band.
 PCR amplification of the EGFP coding region in wells A1 and A2 revealed only a single band approximately 700 bases in length (Figure 10). A band with a similar size was observed for PCR amplification of negative controls pT7GFP and well B2. This result suggests that the CRISPR-Cas12a system did not cut the EGFP coding region on pT7GFP in wells A1 and A2.
PCR amplification of the EGFP coding region in wells A1 and A2 revealed only a single band approximately 700 bases in length (Figure 10). A band with a similar size was observed for PCR amplification of negative controls pT7GFP and well B2. This result suggests that the CRISPR-Cas12a system did not cut the EGFP coding region on pT7GFP in wells A1 and A2.
 PCR amplification of the EGFP ORF on pT7GFP after transfection with a Cas9 and sgRNA-encoding plasmid revealed differences in DNA band sizes.
PCR amplification of the EGFP ORF on pT7GFP after transfection with a Cas9 and sgRNA-encoding plasmid revealed differences in DNA band sizes.
Since there was no clear indication that our CRISPR-Cas12a system was working, we switched to a CRISPR-Cas9 system. HeLa cells were transfected with pT7GFP and one or more pLentiCRISPR-EGFP-sgRNA plasmids, which express human codon-optimized Cas9 protein with an EGFP-targeting synthetic sgRNA (Addgene).pLentiCRISPR-EGFP-sgRNA 1, 2, 4, and 5 targets the EGFP ORF between nucleotides: 28 and 29; 69 and 70; 304 and 305; 358 and 359 respectively. After 24 hours, the cells were collected and the EGFP ORF was amplified via PCR (Figure 11). PCR amplification was performed using EGFP Forward Primer 255, which binds the EGFP ORF starting at nucleotide 255, and EGFP Reverse Primer 420, which binds the EGFP ORF starting at nucleotide 420. For cells transfected with pT7GFP without the Cas9 system, the expected PCR amplicon size was 165 base pairs. For cells transfected with pT7GFP and the pLentiCRISPR-EGFP-sgRNA, CRISPR-Cas9-induced blunt end DNA double-stranded breaks were expected to be repaired via NHEJ, which typically leads to indels. As a result, HeLa cells that expressed both the Cas9 protein and EGFP-targeting sgRNA were predicted to produce a DNA band different in size compared to PCR amplification of pT7GFP without Cas9 or the sgRNA.
 PCR amplification of the EGFP-coding region on pT7GFP revealed one band of varying lengths in each lane (Figure 11). Cells transfected with the full CRISPR-Cas9 system had a larger mean and range of estimated PCR band sizes compared to cells that were not transfected with the system. This is consistent with indel formation during NHEJ, suggesting that pT7GFP was successfully targeted and cleaved by the Cas9 endonuclease and gRNAs.
PCR amplification of the EGFP-coding region on pT7GFP revealed one band of varying lengths in each lane (Figure 11). Cells transfected with the full CRISPR-Cas9 system had a larger mean and range of estimated PCR band sizes compared to cells that were not transfected with the system. This is consistent with indel formation during NHEJ, suggesting that pT7GFP was successfully targeted and cleaved by the Cas9 endonuclease and gRNAs.
PCR amplification of the F13-GFP ORF on a recombinant vaccinia virus after transfection with a Cas9/sgRNA-encoding plasmid revealed differences in DNA band intensity.
It was hypothesized that the CRISPR-Cas12a system showed no clear fragmentation of the VACV genome because the genome was made inaccessible to the CRISPR-Cas12a system by the capsid or replication factory. To determine if the CRISPR-Cas9 system could access the EGFP ORF on the VACV genome, HeLa cells were transfected with pLentiCRISPR-EGFP-sgRNA 2 for 24 hours to allow for the expression of the CRISPR-Cas system off of cellular promoters. SpCas9 endonuclease expression was driven by the elongation factor 1α short (EFS) promoter and gRNAs expression was driven by the RNA polymerase type III U6 promoter. The cells were then infected with vA4mKateF13GFP at an MOI of 3 for 24 hours. pLentiCRISPR-EGFP-sgRNA 2 was specifically chosen because PCR amplification of the EGFP ORF in pT7GFP after pLentiCRISPR-EGFP-sgRNA 2 transfection produced the largest bands compared to the other pLentiCRISPR-EGFP-sgRNA plasmids (Figure 11). The band’s size increase is likely due to nucleotide additions during NHEJ after Cas9-mediated cleavage. Since the efficiency of NHEJ for the VACV genome after Cas9 endonuclease cleavage has been suggested to be an inefficient process, most of the VACV genomes cut by the Cas9 endonuclease should remain fragmented at the EGFP ORF (13). If the EGFP ORF remains fragmented, full-length EGFP mRNA will not be transcribed, reducing EGFP expression. Additionally, PCR amplification using primers that bind upstream and downstream of the cut site will not amplify the segment if it is fragmented. Lastly, it remains uncertain whether the viral genome, when cut approximately in half, will be encapsidated or not. If it is not packaged, fewer progeny virions will be made. If the fragment is packaged, the progeny virions will not effectively infect and replicate inside cells since half of the genome is missing. As a result, it was expected that cells transfected with the Cas9 and gRNA expressing plasmid would have decreased EGFP mean fluorescence intensity (MFI), amplicons following PCR amplification of the EGFP ORF, and decreased infectious progeny virus production after infection with vA4mKateF13GFP.
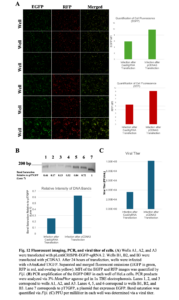
Transfected and infected cells were imaged 24 hours post-infection. Reduced fluorescence for both EGFP and RFP was observed for cells transfected with pLentiCRISPR-EGFP-sgRNA (Wells A1, A2, and A3) compared to cells transfected with pCDNA3 (Wells B1, B2, and B3) (Figure 12A). Further analysis and quantification of the fluorescence intensity revealed that the mean fluorescence intensity was reduced for cells transfected with pLentiCRISPR-EGFP-sgRNA compared to cells transfected with pCDNA3 (Figure 12A). Cells were harvested as before and analyzed by PCR. PCR amplification of the EGFP ORF resulted in fewer amplicons for cells transfected with pLentiCRISPR-EGFP-sgRNA compared to cells transfected with pCDNA3 (Figure 12B). Lastly, cells transfected with pLentiCRISPR-EGFP-sgRNA produced fewer PFU per mL compared to cells transfected with pCDNA3 (Figure 12C).
Discussion:
To explore poxvirus DNA genome replication and packaging, a CRISPR-Cas12a system and CRISPR-Cas9 system were utilized to fragment the viral genome at specific sites. To determine the necessary conditions under which these systems would function, crRNAs targeting the EGFP gene were used, and the impact on EGFP expression was analyzed.
When cells were infected with a recombinant VACV expressing EGFP and then transfected in plasmids expressing the CRISPR-Cas12a system from a viral promoter, there were no observed changes in EGFP expression between the experimental and control groups. The reason for this outcome could be the high MOI used for the infection (Figure 3), as the increased viral genomes in cells during infections at high MOIs can trigger double-stranded DNA sensors that stimulate cytokine expression, which increase cell stress and cell death compared to infections at lower MOIs. Additionally, a high MOI may induce antiviral cellular responses, complicating experimental results. Moreover, the genome’s replication rate may outpace the CRISPR-Cas12a system’s ability to locate and cleave it. For example, if the template genome is fragmented before replication, fewer replicated genomes will be produced. In contrast, if the viral genome is cut after many replication cycles, the number of replicated genomes will be higher. No difference in EGFP expression was observed between the experimental and the control well when different MOIs were used. A viral titer was performed to determine the replication process efficiency and overall success of the initial infection procedure. The CRISPR crRNAs target EGFP sequences on the viral genome for recombinant VACVexpressing EGFP-tagged proteins, which should lead to unpackaged viral genome cleavage and the subsequent reduction of infectious progeny virions. However, the concentration of plaque-forming units in the experimental conditions (CRISPR-Cas12a with crRNAs) was either the same or greater than the control conditions (CRISPR-Cas12a without crRNAs), indicating that the system was not sufficient to reduce viral replication.
Initially, we hypothesized that the failure of FnCas12a to cleave the viral genome was because the Cas endonuclease was not expressed or unable to correctly process the crRNAs. Western blot analysis revealed a band consistent with the size of FnCas12a (Figure 5), and EGFP expression remained similar between the experimental and control groups after the crRNA plasmids underwent restriction digestion. Transitioning from a recombinant vaccinia virus expressing EGFP to transgenic EGFP expression plasmids, we saw similar results, with little variation in the percentage of EGFP-expressing cells between the experimental and control conditions. In both experimental and control samples, PCR amplification of 714 base pairs of the EGFP coding region revealed only a single band of approximately 700 base pairs in length. If Cas12a had cut pT7GFP, it would have resulted in staggered cuts that could be repaired via NHEJ, introducing variable indels. Furthermore, two bands–one band at around 714 base pairs and another band 560 base pairs shorter–were expected if two Cas12a-mediated staggered cuts at different sites were generated simultaneously on the pT7GFP plasmid. This would result in two different staggered ends on the plasmid that could be repaired and annealed together, resulting in the removal of around 560 base pairs of the EGFP ORF. This smaller band was not observed, suggesting that multiple simultaneous Cas12a-mediated cleavages of DNA were not observed in our system (Figure 10). Notably, the PCR amplicon produced from cells transfected with the full CRISPR-Cas12a system appears slightly larger than the PCR amplicons of pT7GFP (Figure 10). While this suggests that additions to the EGFP ORF may have occurred–likely as a result of NHEJ repair after Cas12a-mediated cleavage–the gel used did not have a high enough resolution for us to make any conclusions. Moving forward, nucleotide additions could be verified by sequencing clones from the agarose gel bands.
Our CRISPR-Cas12a system’s ability to cut plasmid DNA but not viral DNA could be explained by the timing of the CRISPR-Cas12a system expression during viral infection. The CRISPR-Cas12a plasmids utilize intermediate VACV gene promoters, and intermediate genes are expressed after viral DNA replication. Upon entry into a cell, the viral genome is surrounded by a proteinaceous shell that later undergoes a poorly understood uncoating process, when the core dissolves and the genome is released (7). Genome replication occurs in discrete cytoplasmic structures called replication factories (7). By the time the Cas12a endonuclease and crRNAs are expressed, the VACV genome is already sequestered in the viral factory and replicating. As a result, the VACV genome may not be accessible to the CRISPR-Cas12a system for cleavage. Efficient VACV genome cleavage was strongly suggested by the results in Figure 12 when cells were transfected with the CRISPR-Cas9 system and then infected with VACV 24 hours later. Transfecting before infecting the cells allows the CRISPR-Cas9 system to be present in the cell before any viral DNA is present. After virion entry and uncoating, the genome localizes to replication factories (7). During this process, it is exposed to the cytoplasm and may be targeted by the CRISPR-Cas9 system.
Another explanation for the observed inefficiency of the CRISPR-Cas12a system could be the transfection protocol. Using 5 µl of Lipofectamine with 0.25 µg of pT7GFP, an EGFP-expressing plasmid, resulted in approximately 20% of the cells fluorescing green. In comparison, using 5 µl of Lipofectamine 2000 with 0.25 µg of the same plasmid led to approximately 70% of the cells fluorescing green. Moving forward, revisiting prior infection and transfection experimental setups and replacing Lipofectamine with Lipofectamine 2000 to improve transfection efficiency may lead to different results. The low transfection efficiency seen with Lipofectamine and pT7GFP indicates that many of the cells in earlier experiments did not receive all the components necessary for the CRISPR-Cas12a system to function properly. After transfection, CRISPR-Cas12a functionality could be determined through counting cells and PCR amplification of the EGFP coding region. Once the optimal conditions for FnCas12a activity are determined, crRNAs for specific areas of the VACV genome can be developed to fragment the genome and explore the mechanisms of poxvirus genome replication and packaging.
Plasmids expressing human codon-optimized Cas9 protein were transfected with EGFP-targeting synthetic sgRNA. Cells are then infected with a verified VACV expressing EGFP-tagged proteins to decrease fluorescence, viral EGFP template for PCR, and progeny virion production compared to the negative control. This strongly suggests the VACV genome was targeted by the Cas9 endonuclease. To further optimize Cas9 endonuclease activity, cells could be transfected with a plasmid expressing a Cas9 that does not possess nuclear localization signals (NLS). NLS is an amino acid motif that mediates protein transport into the nucleus (17), and is often appended to Cas9 endonucleases so the Cas9 protein can be transported into the nucleus, where DNA is present. These NLS may hinder VACV genome cleavage because the VACV genome replicates in the cytoplasm, not the nucleus, and the NLS signal reduces the number of Cas9 endonucleases in the cytoplasm. It would be beneficial for future studies to design a new plasmid for expressing a Cas9 endonuclease without NLS. Moving forward, to minimize variations in background fluorescence, which result from imaging the cells when covered in DMEM, images will be taken with cells covered with PBS or a media without neutral red (which autofluoresces). As well, quantitative PCR can be used to measure the quantity of present DNA more precisely.
It is hypothesized that within the large dsDNA genome of VACV, there may be packaging signals present. It is unclear if these signals are present and where they may be located. To explore the location of packaging signals and VACV genome packaging, a recombinant VACV expressing mKate-tagged A4 proteins, EGFP-tagged thymidine kinase (TK) proteins, and blue fluorescent protein (BFP) tagged F13 proteins will be utilized. Since the TK-GFP ORF lies near the VACV genome center, transfecting cells with pLentiCRISPR-EGFP-sgRNA 2 before infecting them with this virus should fragment the viral genome near the middle. If NHEJ does not occur, this should produce two distinct fragments, with one containing the A4-mKate ORF and the other containing the F13-GFP ORF. If packaging signals are present on one half but not the other, cells infected with progeny virions should fluoresce predominantly in one color, either red or blue. If packaging signals are present in both halves, an equal number of cells infected with progeny virions should fluoresce red and blue. Moreover, by placing the EGFP ORF in different parts of the genome, different VACV genome fragments can be generated after the EGFP ORF has undergone Cas9-mediated cleavage and tested for packaging.
Understanding VACV genome encapsidation is important for several reasons. First, despite smallpox eradication, there are concerns that it may re-emerge from forgotten stocks, de novo synthesis, or melting permafrost resulting from global warming (6). This is especially concerning given the low rates of smallpox vaccination since the 1980s and the low number of smallpox-specific drugs (6). By understanding poxvirus genome packaging, replication, and the proteins involved in those processes, novel drugs that target essential proteins can be developed. Furthermore, many biotechnology products have been developed based on enzymes identified from the vaccinia virus, like the vaccinia capping system for in vitro synthesized RNA and TOPO cloning, which is based on vaccinia DNA topoisomerase (6). Identifying the enzymes involved in replication or encapsidation may lead to novel poxvirus-based biotechnology products. Lastly, poxviruses are being developed as vaccine vectors, oncolytic therapeutics, and gene delivery vehicles (6). By better understanding viral processes like genome replication and encapsidation, more efficient systems can be designed.
In conclusion, the CRISPR-Cas12a system we used did not efficiently cut the EGFP-coding region in our tests. Related to this outcome could be the low transfection rates seen with Lipofectamine, so the decision was made to replace Lipofectamine with Lipofectamine 2000. Furthermore, the Cas12a sgRNAs designed may not be effectively processed and recognized by the Cas12a endonuclease. Lastly, the CRISPR-Cas12a system was expressed using the I1 promoter, an intermediate VACV promoter. Intermediate genes are expressed after viral genome replication, so by the time the CRISPR-Cas12a system is expressed, the viral genome may be sequestered and unavailable for the CRISPR-Cas12a system to cleave. Since there was no clear indication that pT7GFP was being cut by the CRISPR-Cas12a system, we switched to a CRISPR-Cas9 system. With this system, we observed decreased fluorescence, decreased viral EGFP DNA template for PCR, and decreased progeny virion production compared to the negative control, suggesting that the Cas9 endonuclease was able to fragment the VACV genome. By placing the EGFP ORF in specific regions of the VACV genome and using the CRISPR-Cas9 system to target the EGFP ORF, the VACV genome can be fragmented. Fluorescent protein reporters can help to determine which fragments, if any, are packaged. Understanding how these fragments are packaged can provide insight into the location of packaging signals in the VACV genome.
Materials and Methods:
Cells and viruses. HeLa and BSC-40 cell monolayers were grown in DMEM with 8% HyClone Cosmic Calf Serum (Cytiva) at 37 °C with 5% CO2. All viruses were derived from the WR strain of VACV. Recombinant viruses, listed in Table 2, were generated per standard protocols (16)
| Table 2: Recombinant VACV used | |
| Recombinant VACV | Function |
| vA4mKateF13GFP | Makes RFP-tagged core proteins (A4) and EGFP-tagged enveloped proteins (F13). A4 expression utilizes an early VACV promoter. F13 expression utilizes an intermediate VACV promoter. |
| T7 VACV | Utilizes an early VACV promoter (B5) to express T7 RNA polymerase for the T7 expression system. |
Oligonucleotides. Oligonucleotides were purchased from Invitrogen (Table 3).
| Table 3: Oligonucleotides used in cloning and PCR | ||
| Oligonucleotide Name | Sequence (5’-3’) | Purpose |
| I1 promoter | GATATATTTGTATTTAAAAGTTGTTTGGTGAACTTAAATTTCTACTGTGTAGAT | Synthetic intermediate VACV gene promoter |
| EGFP 3’ gRNA Rev | CTGAGCACCCAGTCCGCCCTGAGATCTACAACAGTAGAAATTTAAGTTCACC | Cas12a gRNA that targets the 3’ end of the EGFP gene. Present in p3gRNA. |
| EGFP 5’ gRNA Rev | CCTGGTCGAGCTGGACGGCGACGATCTACAACAGTAGAAATTTAAGTTCACC | Cas12a gRNA that targets the 3’ end of the EGFP gene. Present in p5gRNA. |
| T7 Forward
Primer |
TAATACGACTCACTATAGGG | PCR amplification of the EGFP coding region |
| Sp6
Reverse Primer |
ATTTAGGTGACACTATA | PCR amplification of the EGFP coding region |
| EGFP Forward Primer
7 |
AGCAAGGGCGAGGAGCTGTT | PCR amplification of the EGFP coding region. Binds starting at nucleotide 7 on the EGFP ORF |
| EGFP Reverse Primer
721 |
CTTGTACAGCTCGTCCATGCCG | PCR amplification of the EGFP coding region. Binds starting at nucleotide 721 on the EGFP ORF |
| EGFP Forward Primer
255 |
CTTCAAGTCCGCCATGCCCG | PCR amplification of the EGFP coding region. Binds starting at nucleotide 255 on the EGFP ORF |
| EGFP Reverse Primer
420 |
CCCCAGGATGTTGCCGTCCT | PCR amplification of the EGFP coding region. Binds starting at nucleotide 420 on the EGFP ORF |
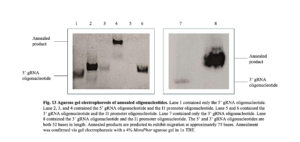
Plasmids. To construct a plasmid that expresses the CRISPR-Cas12a sgRNAs, the I1 promoter oligonucleotide was annealed separately to the 5’ gRNA oligonucleotide and 3’ gRNA oligonucleotide via PCR using OneTaq DNA polymerase, as per “NEB® PCR Cloning Kit” protocol (New England Biolabs). The following PCR cycling parameters were used: 94 °C for 5 minutes, 89 °C for 2 minutes, 84 °C for 2 minutes, 79 °C for 2 minutes, 76 °C for 2 minutes, 68 °C for 5 minutes, and held at 4 °C indefinitely for temporary storage. Annealment was confirmed via gel electrophoresis with a 4% MetaPhor agarose gel in 1x TBE. Unannealed oligonucleotides migrate at approximately 50 bases in length while annealed products migrate at approximately 75 bases (Figure 13). Annealed PCR products were ligated into a linearized pMiniT 2.0 vector (included in “NEB® PCR Cloning Kit”, New England Biolabs). The ligation reaction contained 1 μl of linearized pMiniT 2.0 vector, 2 μl of the insert, 2 μl of H2O, 4 μl of Cloning Mix 1, and 1 μl of Cloning Mix 2. Cloning mixes 1 and 2 provide T4 DNA Ligase, ATP, proprietary ligation enhancers, and end-polishing components. Ligation reaction mixtures were incubated at room temperature (25 °C) for 15 minutes, incubated on ice for 2 minutes, and transformed immediately into NEB 10-beta Competent Escherichia coli (supplied with the kit).
 The transformation protocol called for the thawing of a 50 μl tube of competent cells on ice for 10 minutes, then adding 2 μl of the ligation reaction and flicking the tube 4 to 5 times to mix. The cells were incubated on ice for 20 minutes and then heat-shocked at 42 °C for 30 seconds. Cells were chilled on ice for 5 minutes and then 950 μl of NEB 10-beta/Stable Outgrowth Medium (supplied with the kit) was added. Cells were grown at 37 °C for 60 minutes, shaking at 250 rpm, then plated on agar plates containing 100 μg/ml ampicillin. Agar plates were placed in a 37 °C incubator overnight. The transformation and plating steps stated above were repeated using transformation-competent E. coli and the linearized pMiniT 2.0 plasmid without the ligated product. This acted as a vector-only control. Colony PCR was performed by picking an isolated colony with a wooden applicator and dipping that applicator in a PCR tube. PCR amplification was performed using the T7 forward primer and Sp6 reverse primer. The following PCR cycling parameters were used: initial denaturation at 94 °C for 5 minutes, 35 cycles consisting of denaturation at 94 °C for 30 seconds, annealing at 50 °C for 30 seconds, extension at 72 °C for 30 seconds, then a final extension at 72 °C for 7 minutes, and lastly a hold at 4 °C. The PCR mixture was run on a 2% MetaPhor agarose gel in 1x TBE. The applicator was dipped into a culture tube containing approximately 5 mL Luria-Bertani (LB) liquid media broth containing ampicillin (50 µg/ml). Culture tubes were incubated at 37 °C for approximately 12 to 18 hours with shaking at 250 rpm. Bacteria were pelleted and plasmids were isolated via the miniprep DNA extraction method (Wizard® Plus SV Minipreps DNA Purification System, Promega). Plasmid sequencing via Genewiz confirmed the insert was present in the plasmid.
The transformation protocol called for the thawing of a 50 μl tube of competent cells on ice for 10 minutes, then adding 2 μl of the ligation reaction and flicking the tube 4 to 5 times to mix. The cells were incubated on ice for 20 minutes and then heat-shocked at 42 °C for 30 seconds. Cells were chilled on ice for 5 minutes and then 950 μl of NEB 10-beta/Stable Outgrowth Medium (supplied with the kit) was added. Cells were grown at 37 °C for 60 minutes, shaking at 250 rpm, then plated on agar plates containing 100 μg/ml ampicillin. Agar plates were placed in a 37 °C incubator overnight. The transformation and plating steps stated above were repeated using transformation-competent E. coli and the linearized pMiniT 2.0 plasmid without the ligated product. This acted as a vector-only control. Colony PCR was performed by picking an isolated colony with a wooden applicator and dipping that applicator in a PCR tube. PCR amplification was performed using the T7 forward primer and Sp6 reverse primer. The following PCR cycling parameters were used: initial denaturation at 94 °C for 5 minutes, 35 cycles consisting of denaturation at 94 °C for 30 seconds, annealing at 50 °C for 30 seconds, extension at 72 °C for 30 seconds, then a final extension at 72 °C for 7 minutes, and lastly a hold at 4 °C. The PCR mixture was run on a 2% MetaPhor agarose gel in 1x TBE. The applicator was dipped into a culture tube containing approximately 5 mL Luria-Bertani (LB) liquid media broth containing ampicillin (50 µg/ml). Culture tubes were incubated at 37 °C for approximately 12 to 18 hours with shaking at 250 rpm. Bacteria were pelleted and plasmids were isolated via the miniprep DNA extraction method (Wizard® Plus SV Minipreps DNA Purification System, Promega). Plasmid sequencing via Genewiz confirmed the insert was present in the plasmid.
The plasmids p262, pT7GFP, pCas12a, and p331 were previously constructed in the lab. p262 uses the pCR2.1 vector and makes LF-mCherry-A4-RF. pT7GFP plasmid consists of EGFP inserted between the cytomegalovirus (CMV) and SP6 promoters on pcDNA3. pCas12a utilizes the pCR2.1 vector backbone and makes FnCas12a with a 3’ HA tag using the A34 promoter, a VACV intermediate promoter. p331 utilizes the pGen7 vector backbone and makes the Ebola virus recombinase. pY001 (pFnCpf1_full) was a gift from Feng Zhang (Addgene plasmid # 69973 ; http://n2t.net/addgene:69973 ; RRID:Addgene_69973). pRDA_221 was a gift from John Doench & David Root (Addgene plasmid # 169142 ; http://n2t.net/addgene:169142 ; RRID:Addgene_169142). AsCas12a was a gift from Isaac Hilton (Addgene plasmid # 188492 ; http://n2t.net/addgene:188492 ; RRID:Addgene_188492). LentiCRISPR – EGFP sgRNA 1 was a gift from Feng Zhang (Addgene plasmid # 51760 ; http://n2t.net/addgene:51760 ; RRID:Addgene_51760). LentiCRISPR – EGFP sgRNA 2 was a gift from Feng Zhang (Addgene plasmid # 51761 ; http://n2t.net/addgene:51761 ; RRID:Addgene_51761). LentiCRISPR – EGFP sgRNA 4 was a gift from Feng Zhang (Addgene plasmid # 51763 ; http://n2t.net/addgene:51763 ; RRID:Addgene_51763). LentiCRISPR – EGFP sgRNA 5 was a gift from Feng Zhang (Addgene plasmid # 51764 ; http://n2t.net/addgene:51764 ; RRID:Addgene_51764).
Transfection. HeLa cells were maintained in DMEM with 8% HyClone Cosmic Calf Serum (Cytiva) at 37 °C with 5% CO2. This media was aspirated, and the wells were washed with 1.5 mL of DMEM (-). 800 μl of DMEM (-) was added to each well and each well was transfected with 200 μl of DMEM (-) containing 5 μl of Lipofectamine or Lipofectamine 2000 (Invitrogen) and 1 or 1.25 μg of DNA. Before addition to wells, Lipofectamine and DNA were incubated together at room temperature for 20 minutes. Plates were incubated at 37 °C with 5% CO2 overnight.
Infection and transfection. HeLa cells were maintained in Dulbecco’s modified Eagle medium (DMEM) with 8% HyClone Cosmic Calf Serum (Cytiva) at 37 °C with 5% CO2. For EGFP and RFP expression via a recombinant vaccinia virus, HeLa cells were infected with the virus at an MOI of 3 and concentration of 1 x 108 PFU/mL in 500 μl of DMEM-2 (2% HyClone Cosmic Calf Serum). HeLa cells were infected with vA4mKateF13GFP at an MOI of 3 and concentration of 6 x 107 PFU/mL in 500 μl of DMEM-2 for EGFP expression in infected cells. HeLa cells that were to be transfected with plasmids containing an ORF downstream of a T7 promoter were infected with a T7 VACV.
After aspirating the DMEM-8 (8% HyClone Cosmic Calf Serum) media, monolayers of HeLa cells in 6-well cell culture plates (1.5 x 106 cells per well) were infected with the virus in 500 μl of DMEM-2. 5 μl of 100x araC (working concentration of 40 μg/mL) was added to the wells containing the T7 VACV. Plates were incubated at 37 °C with 5% CO2 for 2 hours and rocked at 20-minute intervals. After these 2 hours, DMEM-2 was aspirated, each well was washed with 1.5 mL of DMEM (-), the DMEM (-) was aspirated, and 800 μl of DMEM (-) was added to each well. Each well was transfected with 200 μl of DMEM (-) containing 5 μl of Lipofectamine or Lipofectamine 2000 (Invitrogen) and 1 μg of DNA. Before addition to wells, Lipofectamine and DNA were incubated together at room temperature for 20 minutes. 10 μl of 100x araC was added to wells containing the T7 VACV. Plates were incubated at 37 °C with 5% CO2 overnight.
Analysis of cells following transfection. Cells were imaged via fluorescence microscopy, and EGFP-expressing cells were manually counted through visual inspection. Quantification of cell fluorescence was performed via Fiji, an image-processing program. Viruses were separated from host cells via centrifugation (18,000 RCF for 5 minutes). Cells were harvested by scraping the cells and the wells were washed with phosphate-buffered saline (PBS). Cell pellets were collected via centrifugation (18,000 RCF for 30 seconds). To determine if the EGFP sequence was cut, plasmids were isolated via the miniprep DNA extraction method (Wizard® Plus SV Minipreps DNA Purification System, Promega). PCR amplification of the EGFP coding region was performed using the T7 primer and Sp6 primer, or custom EGFP primers (Table 2) purchased from Invitrogen. PCR products were analyzed via 1.8% MetaPhor agarose gel in 1x TBE electrophoresis, 3% MetaPhor agarose gel in 1x TBE electrophoresis, or 4% MetaPhor agarose gel in 1x TBE electrophoresis. Agarose band saturation was quantified via Fiji.
Western Blot. Cells utilized in infection/transfection, transfection/infection, or transfection experiments were pelleted by centrifugation (18,000 RCF for 5 minutes). The supernatant was removed and the cells were lysed using modified radioimmunoprecipitation (RIPA) buffer (0.2 mM phenylmethylsulfonyl fluoride, 150 mM NaCl, 0.5% Sodium Deoxycholate, 1% Triton X-100, 0.1% SDS, 10 mM Tris pH 7.4). The lysate and lithium dodecyl sulfate (LDS) sample buffers were loaded on a 4-12% Bis-Tris polyacrylamide gel (Invitrogen) and then ran in an MES buffer. Proteins were then transferred to a nitrocellulose membrane. Blots were incubated with a rabbit anti-HA antibody (Sigma-Aldrich) followed by an anti-rabbit Cy5 antibody (Jackson ImmunoResearch). Blots were visualized via chemiluminescence using the reporter enzyme horseradish peroxidase (HRP).
Restriction enzyme digest. 1 μl of the restriction enzyme, 1 μg of DNA, 5 μl of 10X restriction enzyme buffer (NEBuffer r3.1, New England Biolabs), and 41 μl of H2O were mixed and incubated at 37 °C for one hour. The restriction enzyme was heat-inactivated at 65 °C for 15 minutes.
Titration of virus. BSC-40 cells were maintained in DMEM-10 (10% HyClone Cosmic Calf Serum) at 37 °C with 5% CO2. The media was aspirated and the BSC-40 cells were infected with 500 μl of DMEM-2 containing a virus. Plates were incubated at 37 °C with 5% CO2 for 2 hours and rocked at 20-minute intervals. The media was aspirated and 1.5 mL of DMEM-2 with methylcellulose was added. Cells were incubated at 37 °C with 5% CO2 overnight.
References:
- Chelikani V, Ranjan T, Kondabagil K. 2014. Revisiting the genome packaging in viruses with lessons from the “Giants”. Virology 466-467:15-26.
- Ahi YS, Mittal SK. 2016. Components of Adenovirus Genome Packaging. Front Microbiol 7:1503.
- Tong L, Stow ND. 2010. Analysis of herpes simplex virus type 1 DNA packaging signal mutations in the context of the viral genome. J Virol 84:321-9.
- Fields BN, Knipe DM, Howley PM, Griffin DE. 2001. Fields virology, 4th ed. Lippincott Williams & Wilkins, Philadelphia.
- Moss B. 1990. Regulation of vaccinia virus transcription. Annu Rev Biochem 59:661-88.
- Yang Z, Gray M, Winter L. 2021. Why do poxviruses still matter? Cell Biosci 11:96.
- Greseth MD, Traktman P. 2022. The Life Cycle of the Vaccinia Virus Genome. Annu Rev Virol 9:239-259.
- Grubisha O, Traktman P. 2003. Genetic analysis of the vaccinia virus I6 telomere-binding protein uncovers a key role in genome encapsidation. J Virol 77:10929-42.
- DeMasi J, Du S, Lennon D, Traktman P. 2001. Vaccinia virus telomeres: interaction with the viral I1, I6, and K4 proteins. J Virol 75:10090-105.
- Zetsche B, Gootenberg JS, Abudayyeh OO, Slaymaker IM, Makarova KS, Essletzbichler P, Volz SE, Joung J, van der Oost J, Regev A, Koonin EV, Zhang F. 2015. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 163:759-71.
- Paul B, Montoya G. 2020. CRISPR-Cas12a: Functional overview and applications. Biomed J 43:8-17.
- Wu X, Kriz AJ, Sharp PA. 2014. Target specificity of the CRISPR-Cas9 system. Quant Biol 2:59-70.
- Gowripalan A, Smith S, Stefanovic T, Tscharke DC. 2020. Rapid poxvirus engineering using CRISPR/Cas9 as a selection tool. Commun Biol 3:643.
- Swarts DC, van der Oost J, Jinek M. 2017. Structural Basis for Guide RNA Processing and Seed-Dependent DNA Targeting by CRISPR-Cas12a. Mol Cell 66:221-233 e4.
- Zetsche B, Heidenreich M, Mohanraju P, Fedorova I, Kneppers J, DeGennaro EM, Winblad N, Choudhury SR, Abudayyeh O, Gootenberg JS, Wu WY, Scott DA, Severinov K, van der Oost J, Zhang F. 2017. Multiplex gene editing by CRISPR-Cpf1 using a single crRNA array (vol 35, pg 31, 2017). Nature Biotechnology 35:178-178.
- Earl PL, Moss B. 1991. Generation of recombinant vaccinia viruses, p 16.17.11–16.17.16 In Ausubel FM, et al. (ed), Current protocols in molecular biology, vol 2 Greene Publishing Associates & Wiley Interscience, New York, NY
- Cokol M, Nair R, Rost B. 2000. Finding nuclear localization signals. EMBO Rep 1:411-5.
About the Author
Daniel Kuo graduated from the University of Rochester with a Bachelor of Science in Microbiology. From 2021 to 2024, Dan conducted poxvirus research under the mentorship of Dr. Brian Ward. As an aspiring medical student and physician, Dan hopes to continue his exploration of virology, with a particular focus on oncolytic viruses and viral vectors for gene therapy.
Cite this Article
Kuo., D. and Ward, B. (2024). Exploring Poxvirus Genome Packaging using CRISPR-Cas Systems. University of Rochester, Journal of Undergraduate Research, 23(1). https://doi.org/10.47761/BNHJ9426
JUR | Creative Commons Attribution 4.0 BY International License